As we move from Common Variant Association Studies (CVAS) to Rare Variant Association Studies (RVAS) it has become increasingly obvious that the majority of our computational workload will be dedicated to analyzing rare variants.
For simple illustration here is a side by side barplot showing the absolute number of common variants in a whole genome sequencing study of approximately 200,000 individuals (AllofUs dataset).
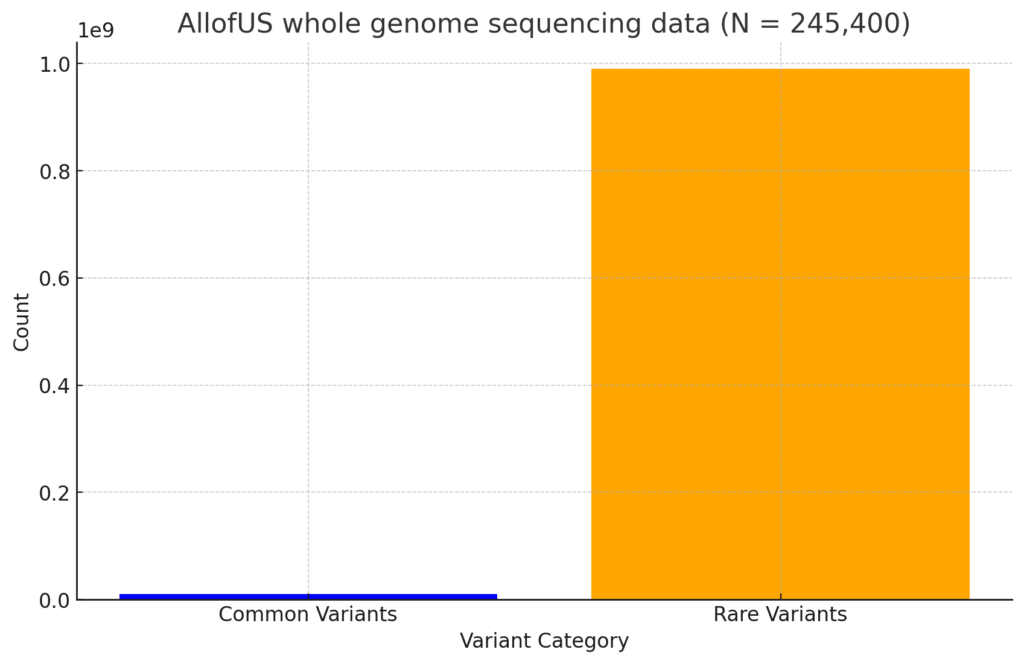
If we were to visualize it in a barplot the difference is quite dramatic.
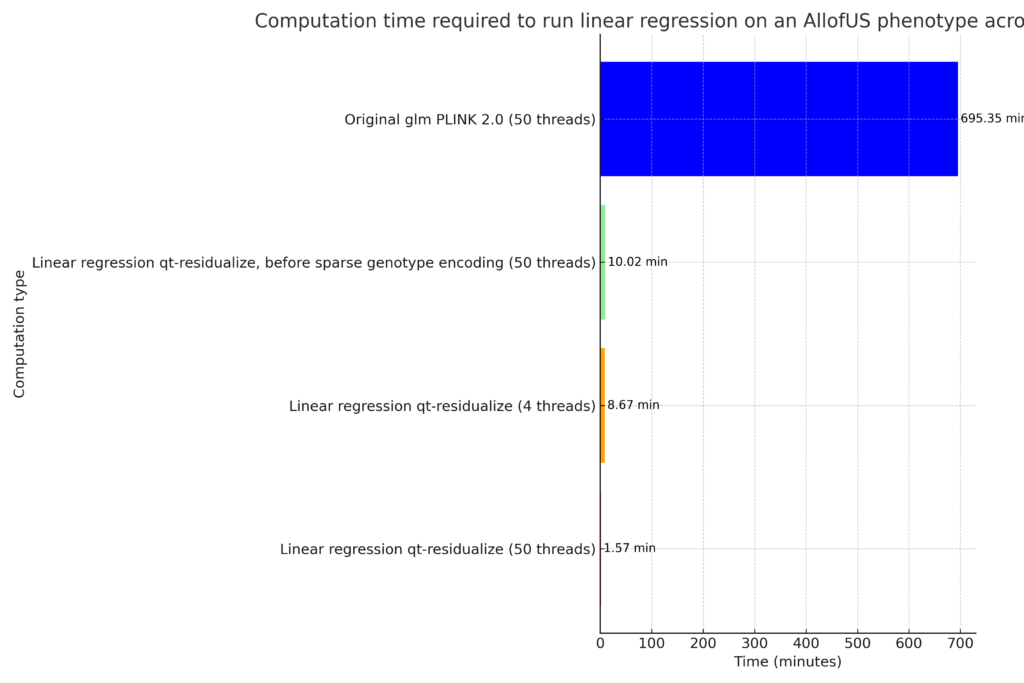
I initially analyzed the body mass index phenotype in the AllofUs cohort using PLINK 2.0.
PLINK 2.0 has computationally efficient algorithms. However, it was quite clear that the original algorithms required quite a lot of computational resources to run it across hundreds of thousands of individuals.
To compare we conducted univariate regression across the exome for the body mass index (BMI) phenotype in the AllofUS cohort, which required 11.6 hours using 50 threads on a single machine.
We realized that we could improve the efficiency of the study by taking advantage of the property that most of the variants that were being analyzed were rare.
To compute the estimates of the regression coefficients we only needed access to the data from the rare variant carriers after residualizing the covariates (this computation only needs to be done once).
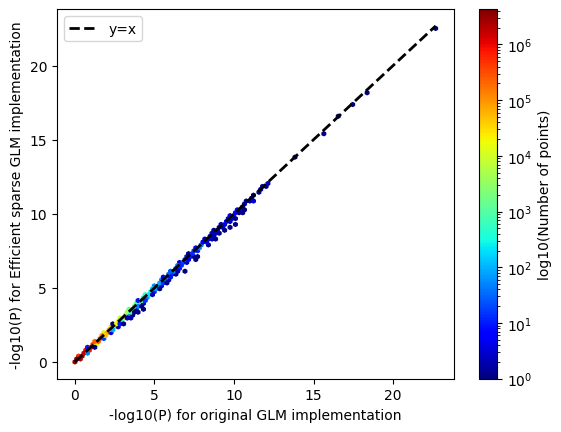
We were able to reduce computation time down from 11.6 hours to 1.6 minutes using 50 threads in a single machine.
We also found that we were able to maintain power and control type 1 error rates.
Paper is found here on biorxiv and it is In Press at Bioinformatics.
Leave a Reply