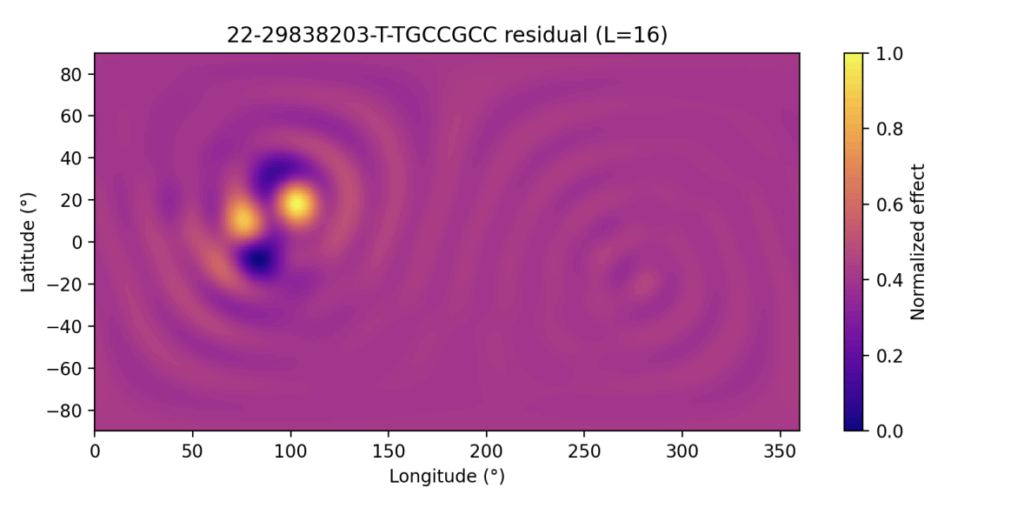
Take home: we introduce a spherical-harmonics regression framework for PheWAS that embeds phenotypes on the unit sphere to jointly model global genetic effect patterns and detect variant-specific, spatially localized signals via residual analysis and spherical-cap enrichment, enabling interpretable, rotation-invariant mapping of genetic effects across phenomes.
Efficient regression for whole genome sequencing studies.
Take home: we introduce a novel data format and regression framework that dramatically accelerates large-scale genomic analyses while minimizing storage and computational costs.
Foundation model pipeline for docking small molecules to genetically validated targets.
Take home: we introduce the Smiles2Dock dataset with ~25M protein–ligand scores from docking 1.7M ChEMBL ligands against 15 AlphaFold proteins, plus an initial Transformer baseline.
Take home: we present methods to improve disease risk prediction and survival modeling in the UK Biobank with multi-omics data, with the greatest gains seen for diseases that have both genetic and metabolic components.
Take home: we systematically evaluate a suite of tree-based and linear machine learning methods—including gradient boosting, random forests, and SNPnet—for genotype-to-phenotype prediction using UK Biobank data, optimizing hyperparameters through multi-objective tuning to balance predictive accuracy and computational efficiency.
The TRB variant rs7458379 influences TRBV4-2 expression, highlighting coordinated genetic effects on T-cell receptor composition in narcolepsy.
Take home: We show that narcolepsy type 1 is an autoimmune disorder driven by specific HLA and T-cell receptor variants that shape T-cell responses to influenza infection or vaccination, leading to targeted destruction of hypocretin-producing neurons.
ALDH2*2 is ssociated with coronary artery disease (CAD) and induces endothelial dysfunction.
Take home: We show that SGLT2 inhibitors such as empagliflozin can reverse endothelial dysfunction caused by the common ALDH2*2 alcohol flushing variant, suggesting a potential preventive therapy for coronary artery disease in affected individuals.
Study designs where common controls could be used.
Take home: we show that using well-matched, rigorously harmonized common control datasets can greatly expand the power of sequencing studies, but only when careful attention is paid to ancestry, technical, and design biases that otherwise risk confounding results.
Take home: We identify multiple rare coding variants associated with Crohn’s disease, notably implicatingATG4C, an autophagy-related cysteine peptidase whose loss-of-function variants significantly increase disease risk, highlighting defective autophagy as a causal mechanism in intestinal inflammation.
Evolution of COVID19 at Stanford University including ancestry.
Take home: We show that integrating viral, host genomic, and clinical data from diverse populations reveals ancestry-specific genetic and biological factors underlying COVID-19 severity.
Sparse reduced rank regression for large-scale regression problems.
Take home: We present a method for performing sparse reduced rank regression for large-scale and ultrahigh-dimensional problems with multiple responses.
Take home: we show that rare genetic variants linked to extreme gene expression improve prediction of complex traits like obesity when integrated into risk models, revealing that incorporating expression outlier–associated variants enhances the accuracy and clinical utility of genetic risk prediction.
Take home: we show that genetic variation strongly influences ascending aortic diameter, and a polygenic score derived from these variants can predict risk for thoracic aortic aneurysm across diverse populations, emphasizing the key causal role of blood pressure in aortic disease.
Genistein modulates cannabinoid receptor (CB1) signaling to counteract Δ⁹-THC–induced inflammation and oxidative stress, reducing atherosclerosis risk through cAMP-PKA–NF-κB pathway regulation.
Take home: we show that marijuana’s active compound Δ⁹-THC promotes vascular inflammation and atherosclerosis through CB1 receptor activation, and the soybean isoflavone genistein acts as a CB1 antagonist that blocks these effects.
Decomposition of genetic associations across multiple population biobanks: Biobank Japan and UK Biobank.
Take home: we show that our approach DeGAS (Decomposotion of Genetic Association Studies) can be applied to summary statistics from multiple biobanks to get disease insights.
Bayesian clustering of genetic variants across their lipid profile.
Take home: we present a new Bayesian mixture model for clustering rare variant genetic effects solely based on summary statistics from univariate regression.
Genetic risk scores for diseases based on a combination of traits.
Take home: we present a framework that models polygenic risk as a combination of latent genetic components, enabling interpretable dissection of individual disease risk into biologically meaningful subtypes without sacrificing predictive accuracy.
Take home:we show that transcriptional response to exercise follows distinct, time-dependent trajectories across muscle and blood, identifies SMAD3 as a central regulator, and uncovers age- and sex-specific molecular adaptations that shape how humans respond to physical activity.
C-index for PRS and clinical risk factors for atrial fibrillation.
Take home: we show that a polygenic risk score (PRS) with the traditional CHA₂DS₂-VASc clinical score modestly but significantly improves prediction of ischemic stroke risk among patients with atrial fibrillation.
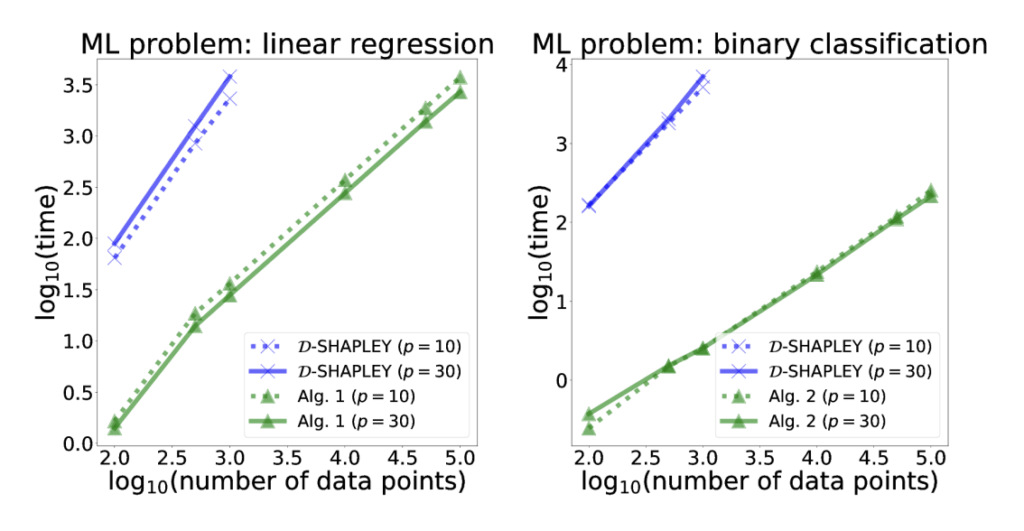
Computation time of our proposed algorithm versus state-of-the art D-Shapley algorithm.
Take home: we derive the first analytic formulas for distributional Shapley values (DShapley)—quantifying each data point’s contribution to model performance—and introduces algorithms that make their computation orders of magnitude faster while offering new theoretical insight into how data characteristics affect value.
ADCY5 genetic variant affects biological processes from the molecular to the organismal level — influencing both glucose metabolism and bone density.
Take home: we find a single noncoding variant, rs56371916 in ADCY5, that links bone and glucose metabolism by altering SREBP1-mediated regulation of lipid oxidation in adipocytes and osteoblasts, revealing a shared genetic mechanism underlying type 2 diabetes and bone mineral density.
GWAS of serum urate identifies key urate transporters and kidney-specific regulatory regions driving urate metabolism.
Take home: we show that even well-understood molecular traits like urate, IGF-1, and testosterone are controlled by a few core biological pathways but also influenced by thousands of small-effect variants across the genome, revealing a highly polygenic genetic architecture.
Association between genetic variant and microbial metabolic pathway in inflammatory bowel disease.
Take home: we show that host genetic variants influence the composition and metabolic functions of the gut microbiota, highlighting key gene–microbiota interactions that shape inflammatory bowel disease pathogenesis.
Take home: we conduct a comprehensive study of 35 blood and urine biomarkers in the UK Biobank that reveals thousands of genetic loci underlying biomarker variation, clarifies causal links to common diseases, and shows that combining biomarker-based polygenic scores improves disease risk prediction beyond single-trait models.
Sex effects mixture model identifies genetic architecture for testosterone levels.Sex specific testosterone polygenic risk score.
Take home: we develop a Bayesian mixture model to model the genetics of testosterone levels in men and females. We also train sex-specific testosterone polygenic risk scores.
snpnet fitting penalized regression to ultrahigh-dimensional problems in population biobanks.
Take home: we introduce BASIL, a fast and scalable algorithm for applying lasso and elastic-net regression to massive, high-dimensional datasets like the UK Biobank, enabling efficient genome-wide prediction and feature selection across millions of genetic variants.
Genetic correlation of predicted risk of suicide and attempted suicide along with other phenotypes.
Take home: we show that common genetic variation contributes meaningfully to suicide attempt risk, and clinically predicted suicide risk from electronic health records shares a significant genetic basis with self-reported suicide attempts.
Tagging clinical notes with ICD codes using deep learning algorithm.
Take home: we present FastTag, a deep learning model that can accurately and efficiently classify unstructured human and veterinary medical narratives into top-level disease categories, reducing the need for manual coding and enabling cross-domain applications.
Type 2 diabetes prevalence increases with higher body mass index (BMI), and this relationship is consistent across individuals with and without a family history of diabetes.
Take home: we show that lower body mass index causally reduces the risk of type 2 diabetes across all levels of genetic risk, family history, and baseline BMI, meaning weight loss benefits everyone—not just those at high risk.
Decomposition of genetic association results reveals biological and biomarker components of disease.
Take home: we show that breaking down genetic associations across thousands of traits using the DeGAs method (SVD of summary statistic matrix) reveals key biological components—particularly those linked to adipocyte biology—that help explain how genetic variation contributes to complex diseases like obesity and heart disease.
TWAS has multiple associations, but susceptible to input data selection.
Take home: we show that there are inherent challenges in transcriptome wide association studies that makes it challenging to identify causal genes by using that approach.
NUDT15 genetic variants associated with thiopurine induced myelosuppression in patients with IBD.
Take home: we identify genetic variants in NUDT15 that associated with thiopurine induced myelosuppression in patients with inflammatory bowel diseases.
Differences in Crohn’s disease genetic risk in Ashkenazi vesus non-Ashkenazi Jewish European population.
Take home: we characterize a founder-effect–driven enrichment of rare and common genetic variants in the Ashkenazi Jewish population contributes significantly to their increased risk of Crohn’s disease and certain rare Mendelian disorders.
Study design of protein-truncating variant phenome-wide association study.
Take home: we conduct a large-scale analysis of protein-truncating variants in over 337,000 UK Biobank participants reveals their significant medical relevance, identifying both protective and risk associations across diverse diseases and underscoring their value for understanding disease mechanisms and therapeutic targeting.
Take home: we reveal that X chromosome inactivation in humans is incomplete and variable across genes, individuals, tissues, and cells, contributing to sex differences in gene expression and potentially influencing health and disease.
Comprehensive transcriptome analysis of protein-truncating variants in GTEx and Geuvadis.
Take home: we present a survey of the effect of protein-truncating variants on the human transcriptome across multiple tissues. We present a model for assessing the effects of variants proximal to splice junctions.
CARD9 IVS11+1G>C protective variant against Crohn’s disease and ulcerative colitis.
Take home: we present a rare exon sequencing study in inflammatory bowel diseases, identify risk and protective mutations in NOD2, CARD9. We develop new methods and software, referred to as Syzygy, for pooled resequencing studies incorporating new error models.
Drug response correlated with growth rate and baseline ATP levels of the cell line.
Take home: we show that genetic variation significantly influences cellular responses to drugs and gene expression levels in vitro, establishing lymphoblastoid cell lines as a powerful model for linking genotype to cellular phenotype.

























 speedup.
speedup.